On going projects
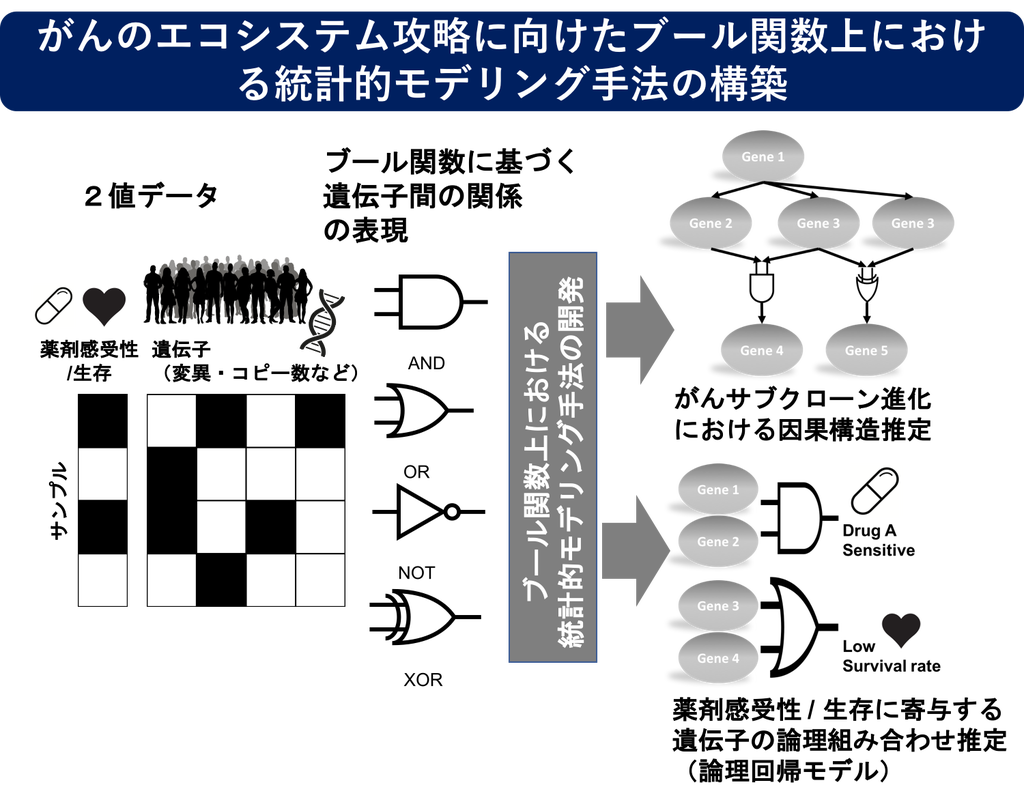
A Statistical Modeling Method on Boolean Functions for Conquering the Cancer Ecosystem
Cancer genome research has accumulated large-scale heterogeneous big data, and recent collaboration between mathematical and computer sciences and life sciences has revealed that cancer autonomously constructs a complex ecosystem among cell populations and weaves a complex life system network beyond human knowledge. In the development of cancer-targeted therapies, it is expected to construct higher-order therapeutic strategies that incorporate the cancer ecosystem, and mathematical modeling methods that enable verifiable and actionable identification of molecular targets are becoming important in addition to conventional comprehensive analysis. In this study, we will develop a squid pustule to capture the ultra-complex cancer ecosystem.
1) mathematical modeling and ultra-fast estimation methods for cancer subclone evolution estimation
2) Robust estimation method for identifying synergistic gene interactions that contribute to drug and survival
1) mathematical modeling and ultra-fast estimation methods for cancer subclone evolution estimation
2) Robust estimation method for identifying synergistic gene interactions that contribute to drug and survival
Completed projects
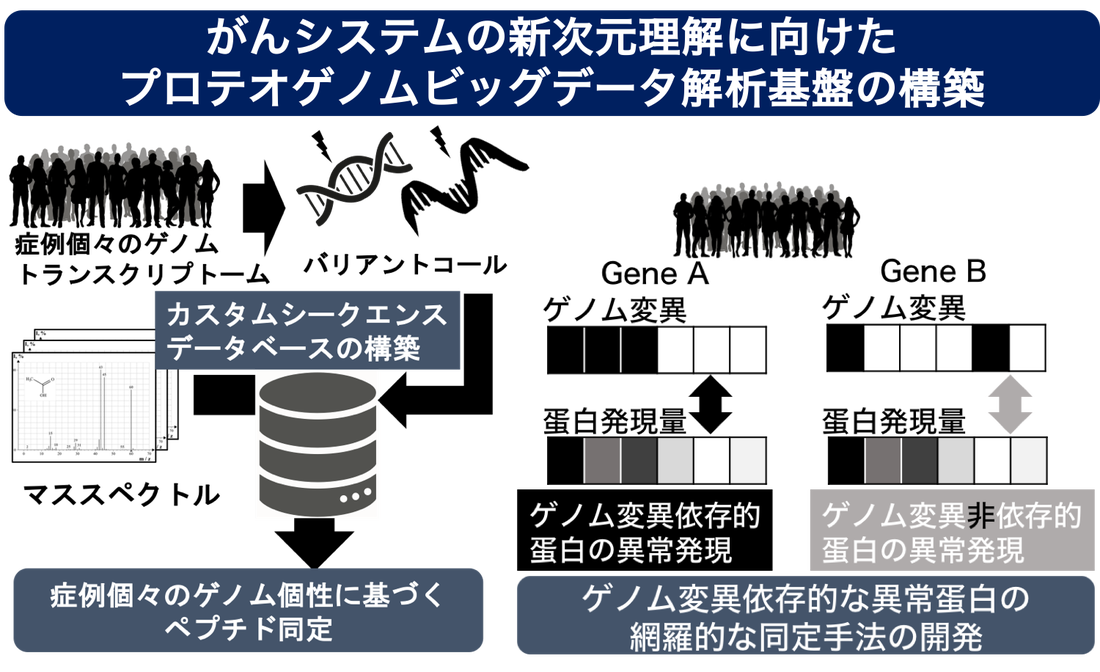
Development of Proteogenomic Platform for Understanding New Dimensions of Cancer Systems
In addition to large-scale cancer genome studies, recent dramatic advances in liquid chromatography-mass spectrometry (LC-MS/MS) have made it possible to acquire large-scale proteome and post-translational modification data on cancer tissues with high throughput. In this research project, we will focus on the proteomic analysis of cancer tissues in order to understand the mechanism of post-translational modification and proteomic changes that define the phenotype of cancer. In this research project, we will establish a large-scale proteogenomic data analysis infrastructure ahead of any other project in Japan by bringing together experts in proteome analysis, genome analysis, and clinical application of proteomics. In particular, we will focus on the following three points. 1.
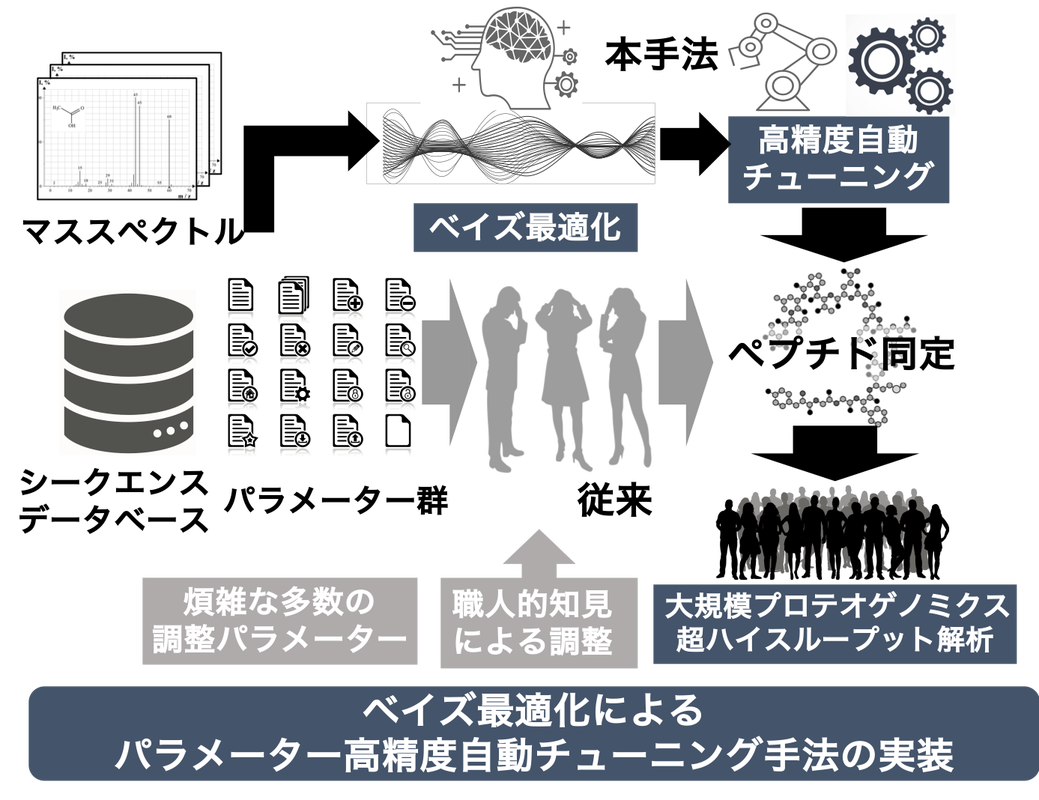
(1) Establishment of an analysis pipeline infrastructure for massively parallel and highly accurate analysis of cancer proteome big data.
2) Development of statistical modeling methods to identify genomic factors that characterize protein and post-translational modifications.
Development of rapid and verifiable predictive models of target molecules based on cell line proteogenomes
(1) Establishment of an analysis pipeline infrastructure for massively parallel and highly accurate analysis of cancer proteome big data.
2) Development of statistical modeling methods to identify genomic factors that characterize protein and post-translational modifications.
Development of rapid and verifiable predictive models of target molecules based on cell line proteogenomes
Development of an Object-Oriented Data Analysis Method for Cancer Diversity
In recent years, the development of mass spectrometry and image analysis, including next-generation sequencers, has led to the accumulation of huge and heterogeneous cancer big data, and the technological basis for their analysis has become an issue. On the other hand, the field of statistical science is also undergoing a data-centered turn, and in addition to the conventional numerical matrix data in multivariate analysis, object-oriented data analysis methods, in which each observed value is represented by a variety of data such as histograms, functions, tree structures, and images, are spreading. Object-oriented data analysis methods are becoming more and more popular. Therefore, we are constructing an infrastructure for object-oriented data analysis in order to exploit a variety of big data in the cancer ecosystem, which is extremely complex .
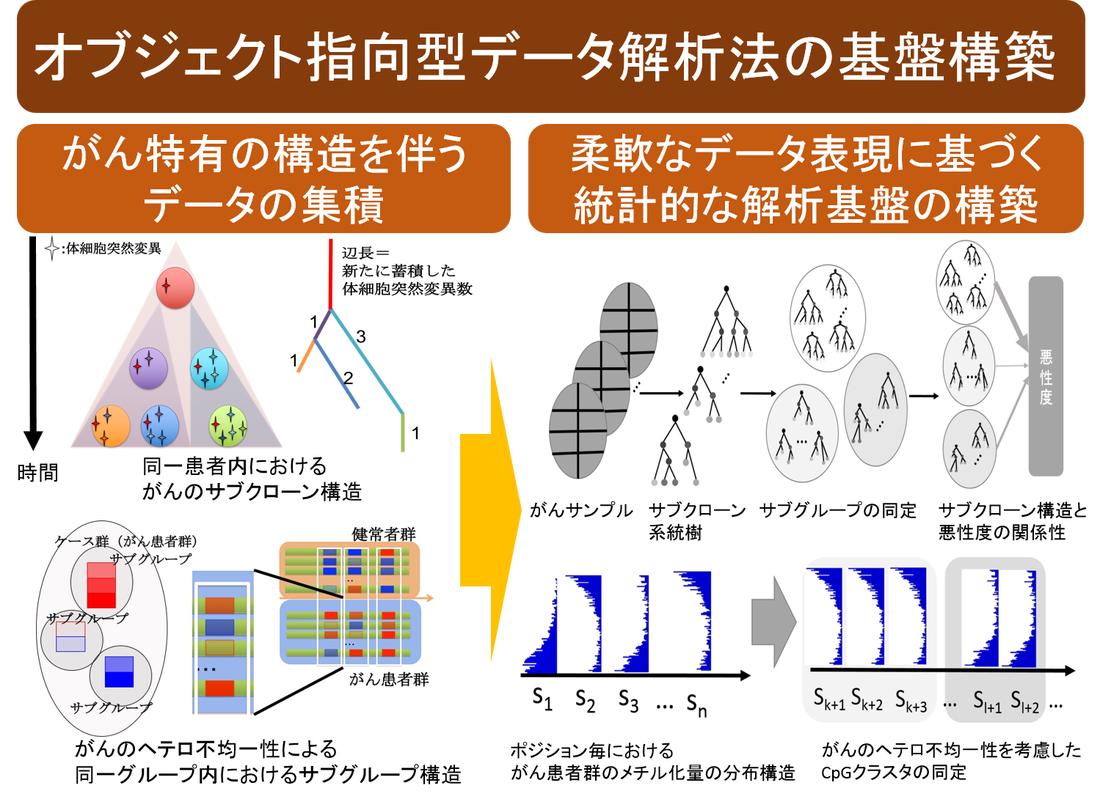
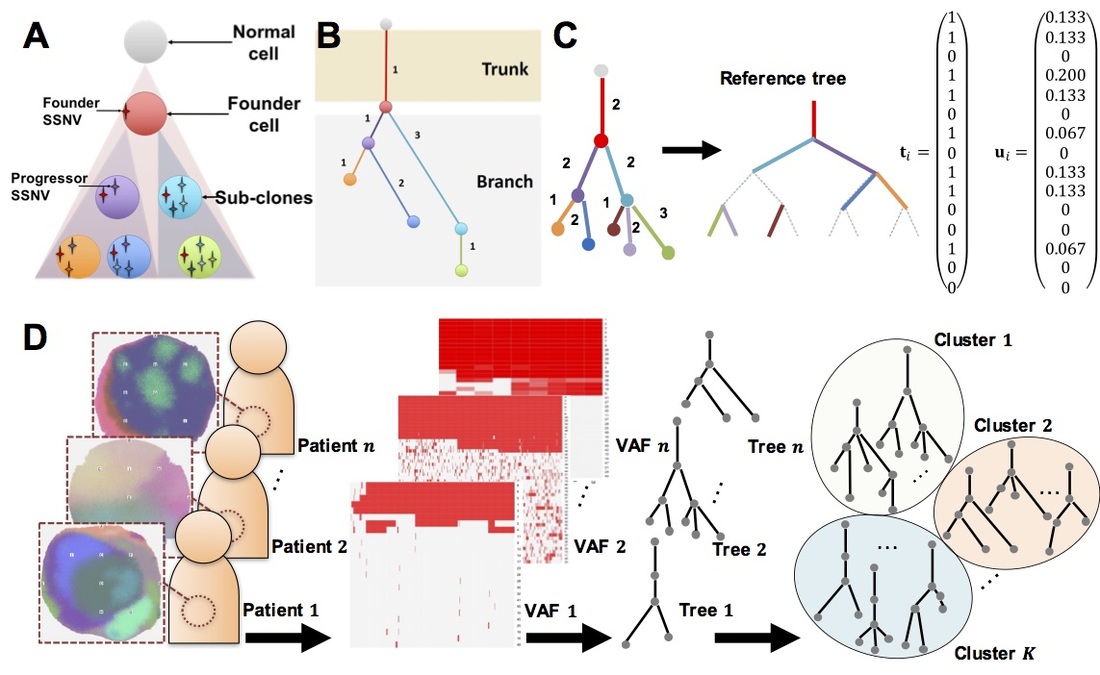
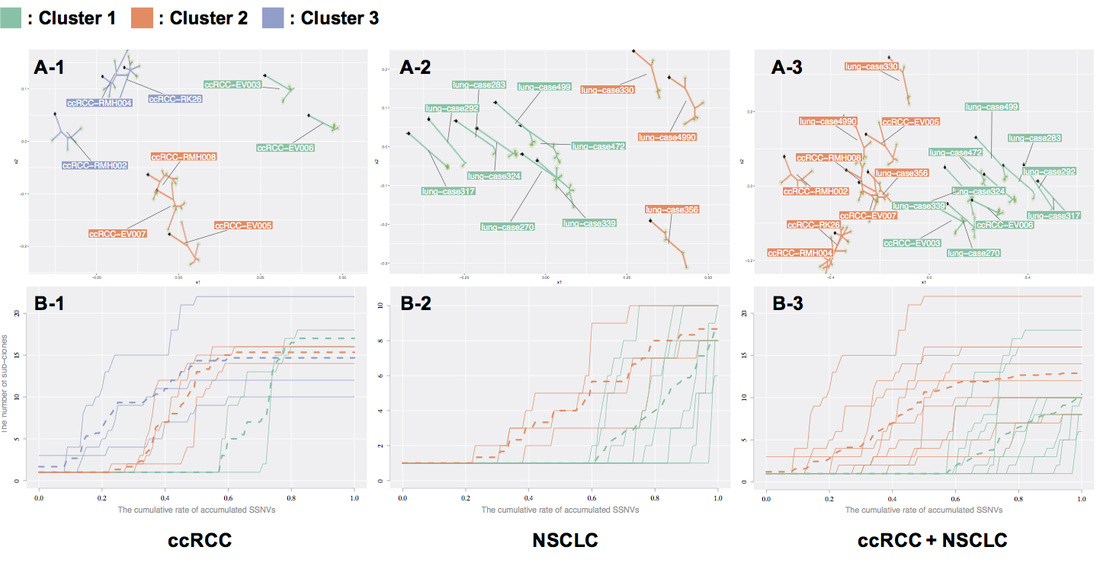
Elucidating cancer subclone structure by tree valued data analysis method
Cancer is thought to be caused by the evolution and abnormal growth of a single normal cell while accumulating genetic mutations. The combination of genetic mutations varies from patient to patient (inter-tumor heterogeneity), and even within a single patient, it has been shown that there are cell populations called sub-clones that have different combinations of genetic mutations (intratumor heterogeneity). It is believed that the resistance and recurrence of cancer are partly due to some resistant subclones in the same cancer, and that resistant subclones, which were originally few in number, acquire resistance by evolving and proliferating to adapt to the changing environment of drug administration. Therefore, it is important in cancer therapy to understand how the structure of subclone evolution differs among cancer cells with different treatment backgrounds. In this study, we have developed an analysis method to estimate the process of subclone evolution in a single cancer sample by examining genetic mutations at multiple different sites using a next-generation sequencer, and to cluster the target groups based on the similarity of subclone evolution by quantifying the differences in subclone evolution obtained from many samples. By quantifying the differences in subclone evolution obtained from a large number of samples, we have developed an analysis method for clustering target groups based on similarities in subclone evolution.
Development of analysis methods for environmental impact assessment data and real-time monitoring data of the Fukushima Daiichi Nuclear Power Plant accident
Monitoring information of environmental radioactivity level [link]
